
Нетифоидные сальмонеллы лидируют в этиологии спорадической и групповой заболеваемости кишечными инфекциями в мире [1, 2]. Среди известных на сегодняшний день более 2500 серотипов сальмонелл доминирующим этиологическим агентом заболеваемости как в мире, так и в России является Salmonella enterica subsp. enterica серовар Enteritidis [3, 4].
Основной причиной быстрого и масштабного распространения S. Enteritidis и укоренения в птицеводческих хозяйствах по всему миру стал международный экспорт из США и ряда европейских стран живой птицы и племенных куриных яиц, контаминированных сальмонеллой [5]. В 1980-е годы сообщалось о высокой частоте выявления серотипа S. Enteritidis в Северной и Южной Америке, Европе и на территории бывшего СССР, а в конце 1980-х и начале 1990-х годов этот патоген начал доминировать в Азии и Африке [6]. После ряда мероприятий, направленных на борьбу с сальмонеллезом в сельском хозяйстве, число случаев заболевания, вызванного S. Enteritidis, во многих странах, включая Великобританию и США, начало снижаться [7]. Но несмотря на принятые меры, вспышки сальмонеллеза, вызванные S. Enteritidis, по-прежнему остаются одними из самых частых во многих странах. Они проявляются в виде массовых эпидемических очагов или проходят под видом повышения уровня спорадической заболеваемости на территориях нескольких городов, регионов и даже стран. Наглядным примером является длительная по времени и масштабная по распространенности вспышка сальмонеллеза S. Enteritidis, которая произошла на территории 16 стран Европейского Союза и привела к 1209 случаям заболевания сальмонеллезом в период с 2015 по 2018 гг. [8].
С генетической точки зрения S. Enteritidis представляет собой один из наиболее гомогенных серотипов Salmonella, из-за чего некоторые клональные линии невозможно дифференцировать с помощью традиционных методов субтипирования, таких как PFGE, MLVA [9]. В предыдущих исследованиях было показано, что большинство изолятов S. Enteritidis принадлежат к одному и тому же доминирующему PFGE-генотипу, обозначаемому на территории Российской Федерации как JEGX01.0001, а на территории США как JEGX01.0004 [10]. Изоляты этого клона наиболее часто обнаруживают при случаях групповой заболеваемости и часто ассоциируют с употреблением мяса птицы и куриных яиц. По данным референс-центра по мониторингу за сальмонеллезами, доминирование этого субтипа среди множества очагов групповой заболеваемости сохраняется в течение последних 10 лет, а это, в свою очередь, не позволяет генетически дифференцировать отдельные эпидемические вспышки и достоверно установить связь с предполагаемыми источниками и факторами распространения инфекции. Использование полногеномного секвенирования, охватывающего все генетические вариации геномов, относящихся к высококлональной линии S. Enteritidis, уже показало свое преимущество при расследовании пищевых вспышек [5, 8, 9]. Возможность точного сопоставления и определения степени генетической близости изолятов, выделенных от пострадавших, предполагаемых факторов передачи инфекции и источников на основе геномных данных, позволяет сформировать доказательную основу для проведения эффективного эпидемиологического анализа.
В настоящее время в Российской Федерации отсутствуют данные об особенностях популяционной структуры изолятов S. Enteritidis, обнаруженных при различных эпидемических ситуациях, на основе геномных данных. В связи с этим цель настоящего исследования заключалась в изучении генетического разнообразия циркулирующих изолятов Salmonella Enteritidis, изолированных при случаях групповой и спорадической заболеваемости в России, с использованием результатов полногеномного секвенирования.
Материалы и методы
Коллекция изолятов S. Enteritidis
Всего в исследовании было проанализировано 460 изолятов S. Enteritidis, полученных в ходе работы референс-центра по мониторингу за сальмонеллезами. Из 460 изолятов 388 выделены от пострадавших людей и предполагаемого источника инфекции и были ассоциированы с 232 очагами групповой заболеваемости сальмонеллезами, а 72 изолята относились к единичным случаям заболеваний или случаям изоляции сальмонелл из продуктов питания и объектов окружающей среды, не связанных со вспышками сальмонеллеза.
Бактериальные изоляты и идентификация
Перед подготовкой ДНК-библиотек для полногеномного секвенирования полученные изоляты рассевали до единичных колоний на среде МакКонки и использовали отдельную колонию для повторной идентификации и серотипирования. Идентификацию каждого изолята на уровне рода Salmonella spp. проводили с помощью системы MALDI Biotyper (Bruker, Германия). В дальнейшем каждый изолят подвергали серологической характеристике по схеме Кауфмана–Уайта. Для серотипирования изолятов сальмонелл использовали поли- («ПЕТСАЛ», Россия) и моноклональные антитела (Sifin, Германия).
PFGE-генотипирование
Для всех изолятов S. Enteritidis поступающих в референс-центр, параллельно проводили субвидовое типирование методом PFGE согласно международному стандартизированному протоколу с использованием фермента рестрикции XbaI [11].
Полногеномное секвенирование
Тотальную ДНК из 7×109 КОЕ экстрагировали с использованием набора DNeasy Blood & Tissue Kit (Qiagen, Германия) или «Рибо-преп» («Амплисенс», Россия). Геномные библиотеки для полногеномного секвенирования каждого изолята сальмонелл готовили из 70 нг тотальной ДНК с использованием набора ДНК-библиотек NexteraXT (Illumina, США). Массовое параллельное секвенирование проводили на платформах MiSeq, Hiseq, NextSeq (Illumina, США).
Обработка данных NGS и оценка качества данных
Удаление адаптеров из нуклеотидных прочтений низкого качества проводили с использованием пакета BBTools (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/). Количество сгенерированных коротких прочтений считалось достаточным, если была достигнута 60-кратная глубина секвенирования не менее 95% длины хромосомы референс-генома Salmonella Enteritidis P125109. Геном каждого изолята был собран de novo с использованием программы Spades v3.12.0 [12] с последующим анализом контигов на предмет глубины секвенирования и таксономической принадлежности к роду Salmonella. Полученные контиги также были использованы для подтверждения принадлежности секвенированного изолята к серотипу S. Enteritidis c помощью программы SISTR [13].
Идентификация однонуклеотидных полиморфизмов и филогенетический анализ
Для реконструкции филогенетического дерева был проведен поиск информативных нуклеотидных вариаций (НВ) путем анализа исходных нуклеотидных прочтений каждого из секвенированных бактериальных изолятов с использованием программного конвейера Snippy v4.5 (https://github.com/tseemann/snippy). В качестве референс-генома использовали нуклеотидную последовательность хромосомы Salmonella Enteritidis P125109. Дополнительно в исследование были включены геном S. Gallinarum str. 287/91 (AM933173) в качестве внешней филогенетической группы для укоренения филогенетического дерева; геномы S. Enteritidis, которые являются представителями трех глобальных филогенетических линий с соответствующими идентификаторами в архиве коротких прочтений NCBI: Атлантическая – ERR3843525, SRR3312300, SRR1968931; Глобальная–ERR3843675, ERR036138, ERR4338350, ERR3864747 и филогенетическая линия США – ERR3864655 [5]. Всего в выборку для филогенетического анализа входило 469 геномов. Полученные профили НВ были конвертированы в формат выравнивания для анализа в программе Gubbins v3.2.1 [14] с последующим анализом вариаций, расположенных вне областей рекомбинаций. Филогенетическую реконструкцию проводили с использованием RAxML v8.2.12 с помощью модели нуклеотидных замен GTR+G. Для подтверждения топологии дерева проводили бутстреп анализ с 1000 повторениями.
Для определения филогенетических сублиний внутри трех глобальных линий мы использовали программу Fastbaps с параметрами optimised symmetric [15].
В дополнение к филогенетическому анализу мы провели группировку изолятов на основании различий в пределах 5 НВ (t5-кластеризация) с помощью программы SKA (Split Kmer Analysis) (https://github.com/simonrharris/SKA) с параметрами по умолчанию. В качестве исходных данных для программы SKA были использованы нуклеотидные прочтения каждого из секвенированных геномов.
Результаты
Филогенетическая кластеризация изолятов и структура популяции S. Enteritidis
Чтобы описать разнообразие циркулирующих сальмонелл и оценить характер группировки изолятов, мы провели кластеризацию данных полногеномного секвенирования, применяя 3 подхода. Первый заключался в реконструкции филогенетического дерева методом максимального правдоподобия, с помощью которого нам удалось понять общий характер группировки исследуемых изолятов и их принадлежность к ранее описанным глобальным филогенетическим линиям S. Enteritidis. Затем определили генетически однородные группы изолятов (филогенетические сублинии) внутри глобальных филогенетических линий с помощью подхода, реализованного в программе Fastbaps. Третий подход заключался в кластеризации исследуемых изолятов на основе различий в 5 НВ, так называемая t5-кластеризация с целью определения генетических групп изолятов, предположительно имеющих эпидемиологическую связь [16].
Всего в исследовании было проанализировано 460 изолятов S. Enteritidis, полученных в ходе работы референс-центра по мониторингу за сальмонеллезами. Изоляты были направлены из 133 населенных пунктов 60 регионов Российской Федерации в период с 2011 по 2022 гг. (рис. 1, см. на вклейке).
Анализ матрицы НВ 469 изолятов путем последовательного сравнения нуклеотидных прочтений каждого из них с референс-геномом позволил выявить 8497 позиций, содержащих НВ, и реконструировать филогенетическое дерево (рис. 2, см. на вклейке). Характер филогенетической кластеризации исследуемых изолятов и изолятов из референтного набора позволил определить принадлежность исследуемых изолятов S. Enteritidis к ранее описанным глобальным филогенетическим линиям: наибольшее число изолятов (n = 408) относились к Глобальной филогенетической линии, 51 изолят – к Атлантической и только 1 изолят – к линии США. При последующем анализе было определено 8 филогенетических сублиний. 7 филогенетических сублиний (GLB_RU 1-7) являлись представителями Глобальной линии, одна сублиния полностью соответствовала Атлантической линии. Из представителей Глобальной линии только сублиния GLB_RU5 была представлена одним изолятом, который был изолирован при спорадическом случае сальмонеллеза в г. Нерюнгри в 2022 г. (рис. 2, см. на вклейке; табл. 1).
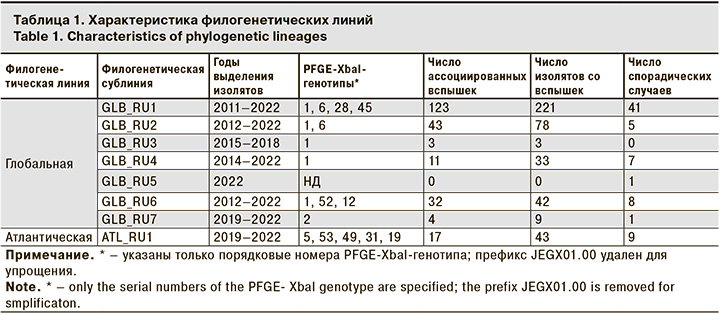
В табл. 1 отражены характеристики выявленных филогенетических линий по дате изоляции, спектру представленности PFGE-генотипов и эпидемической ситуации. Согласно имеющимся метаданным для исследуемых изолятов мы не увидели явной корреляции между филогенетической кластеризацией на уровне филогенетических сублиний и такими параметрами, как источник изоляции и эпидемическая ситуация.
Aнализ филогенетической кластеризации изолятов, характеризующихся PFGE-генотипом JEGX01.0001, показал, что группировка по PFGE-XbaI-генотипу явно коррелирует на уровне глобальных филогенетических линий. Для Глобальной линии и нескольких ее сублиний (GLB_RU 1–6) характерен PFGE-генотип JEGX01.0001. Вместе с тем для 12 (3,5%) изолятов Глобальной линии были характерны PFGE-профили, не относящиеся к JEGX01.0001, что, вероятно, может быть связано с отличающимся спектром плазмид. Филогенетические сублинии ATL_RU1 и GLB_RU7 характеризовались отличным от JEGX01.0001 PFGE-профилем.
Для GLB_RU7 был характерен профиль JEGX01.002, а для сублинии ATL_RU1 – генотипы JEGX01.0005, JEGX01.0019, JEGX01.0053, JEGX01.0049 и JEGX01.0031. Нужно отметить, что выборка изолятов для полногеномного секвенирования была сформирована с приоритетом детального изучения доминирующего PFGE-XbaI-генотипа JEGX01.0001, а сами изоляты были выделены в широком временном диапазоне – с 2011 по 2022 гг. Напротив, изоляты с отличающимися PFGE-генотипами преимущественно были изолированы в период 2019 – 2022 гг.
Степень генетической схожести изолятов внутри очагов групповой заболеваемости сальмонеллезом
Важной задачей являлась оценка эпидемиологической конкордантности между группировкой изолятов по принадлежности к отдельному очагу заболеваемости и характером кластеризации геномов изолятов на уровне различий в 5 НВ. Для ее решения мы оценили принадлежность изолятов, относящихся к случаям групповой заболеваемости, к определенным t5-кластерам. Сравниваемая выборка включала 388 изолятов сальмонелл, полученных из 232 очагов групповой заболеваемости, где 150 случаев (64,6% очагов) было представлено одиночным изолятом, а для 82 (35,4% очагов) было отобрано 2 и более изолята на очаг (49 очагов – 2 изолята, 16 – 3, а для 17 очагов число изолятов варьировало от 4 до 13). В 57 (24,6%) очагах, помимо репрезентативных изолятов от людей, в выборке присутствовали изоляты от вероятного фактора передачи – предполагаемых продуктов и образцов окружающей среды, отобранных в ходе эпидемиологического расследования вспышки.
Общий анализ топологии филогенетического дерева не выявил несоответствий между филогенетической кластеризацией и группировкой изолятов на основании различий в пределах 5 НВ t5-кластеризацией. Среди 213 обнаруженных t5-кластеров число уникальных t5-кластеров составило 171 (80,3%), а в 19 (19,7%) случаях было показано несовпадение t5-кластера изолятов, выделенных из вероятного источника инфекции или от людей.
Последующий анализ очагов с предполагаемым источником инфекции показал, что в 43 (75,4%) очагах из них изоляты от людей и/или из предполагаемых продуктов характеризовались сходным t5-генотипом, что подтверждало эпидемиологическую связь между источником инфекции и пострадавшими людьми. Однако в 14 случаях 1 или более изолятов из продуктов питания не соответствовали доминирующему внутри вспышки t5-генотипу, а в 5 случаях генотипы между изолятами от пострадавших людей не соответствовали друг другу.
Таким образом, полученные данные свидетельствуют о высокой степени соответствия кластеризации геномных профилей (t5-кластеризация) с эпидемиологическими данными и показывают высокую разрешающую способность субтипирования S. Enteritidis на основе геномных данных.
Пространственно-временные особенности кластеризации изолятов, относящихся к эпидемиологически не связанным случаям групповой заболеваемости
Анализ характера кластеризации на уровне различий в 5 НВ всех 460 изолятов (из очагов и спорадические случаи) позволил определить 241 t5-кластер. Классификация кластеров по признаку эпидемиологического фона, при котором был выделен изолят, и уникальности полученного генотипа на уровне 5 НВ позволила выявить 33 кластера, к которым относились спорадические изоляты с уникальным t5-генотипом (45,8% от общего числа спорадических изолятов), и 161 уникальный t5-кластер с 236 (60,8%) изолятами, выделенными при вспышках (табл. 2).

Наиболее интересным с эпидемиологической точки зрения был факт выявления уникальных t5-кластеров, к каждому из которых относились либо более одного спорадического случая, либо изоляты из эпидемиологически не связанных между собой очагов групповой заболеваемости, произошедших в разное время на разных территориях. Было обнаружено 6 t5-кластеров с 13 (18,0%) изолятами, к которым относились спорадические случаи сальмонеллеза или случаи обнаружения сальмонелл из объектов окружающей среды, и 25 t5-кластеров со 100 изолятами (25,7% от всех изолятов из очагов), где каждый кластер включал изоляты, выявленные при разных случаях групповой заболеваемости. Помимо этого, было обнаружено 16 t5-кластеров с 78 изолятами, где каждый кластер включал изоляты, выделенные как во время вспышек, так и при спорадических случаях заболеваний или единичных случаях выделения из объектов окружающей среды – 52 (13,4%), и 26 (36,1%) изолятов соответственно (см. табл. 2). Таким образом, доля изолятов, характеризующихся сходным генотипом, среди циркулирующих спорадических изолятов составил 54,1%, а среди вспышечных – 39,1%.
Сопоставление времени изоляции и географических координат случаев заболеваемости 16 t5-кластеров, включающих и спорадические, и вспышечные изоляты сальмонелл, и 25 t-5 кластеров с изолятами, ассоциированными только с эпидемическими очагами, показало, что временной диапазон обнаружения для кластеров варьировал от 0 до 111 мес. и в среднем составил 18 мес. Вариация максимального значения в попарных географических расстояниях между случаями выделения изолятов внутри каждого t5-кластера, составило от 0 до 4531 км, в среднем 931 км. При этом число t5-кластеров в каждой филогенетической сублинии было следующим: GLB_RU1 – 23, GLB_RU2 – 11, ATL_RU1 – 6, GLB_RU6 – 1.
Максимальное число очагов групповой заболеваемости (6), изоляты которых показали принадлежность к одному генетическому кластеру t5:23, происходили в городах Улан-Удэ, Усть-Илимск, Чита, Красноярск, Биробиджан, Тас-Юрях и 1 случай спорадический заболеваемости в городе Тайшет. Циркуляцию этого t5-генотипа фиксировали на протяжении 4 лет (47 мес.) с декабря 2015 г. по ноябрь 2019 г. При этом максимальная географическая удаленность между очагами групповой заболеваемости составила 2797 км (табл. 3). Филогенетический анализ коррелировал с данными кластеризации на основе различий в НВ. Изоляты t5:23 относились к уникальной филогенетической кладе с уровнем бутстрап поддержки 97%.
Обратная ситуация сложилась для кластера t5:197, к которому относились 6 изолятов S. Enteritidis, при этом 5 из них были выделены при спорадических случаях из продуктов питания в период с 2019 по 2021 гг. в Нерюнгринском районе и городах Нерюнгри, Усолье, Саянск и Богородицы, и только 1 изолят представлял очаг групповой заболеваемости в г. Вилюйск в январе 2019 г. (табл. 3).
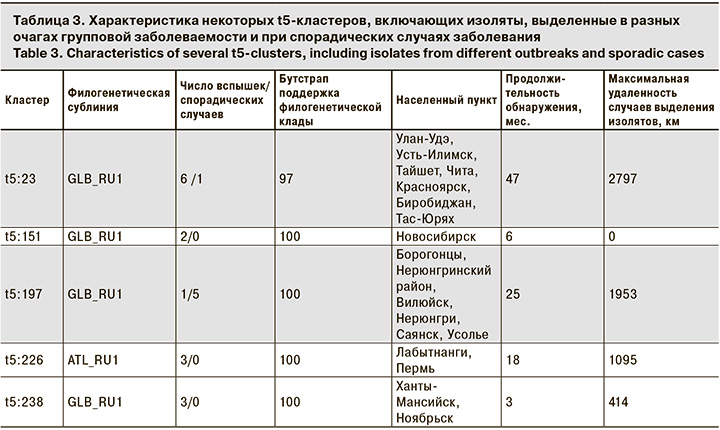
Для ряда t5-кластеров S. Enteritidis был отмечено такое явление, как повторные случаи групповой заболеваемости, вызванные тем же клоном бактерии, спустя некоторое время. Например, вспышки сальмонеллеза, вызванные генотипом t5:226, были зафиксированы в Перми в ноябре 2019 г. и феврале 2020 г.; генотип t5:238 – в Ноябрьске в октябре 2019 г. и декабре 2019 г. и генотип t5:151 – в Новосибирске в сентябре 2016 г. и в марте 2017 г., тем самым иллюстрируя длительный процесс сохранения активности источников одного и того же эпидемического клона S. Enteritidis в пределах определенного населенного пункта.
Обсуждение
В исследовании представлены результаты ретроспективного изучения изолятов S. Enteritidis, циркулирующих на территории России, для описания популяционной структуры и определения филогенетического положения изолятов S. Enteritidis в контексте глобального разнообразия; отражения конкордантности эпидемиологических данных (группировка изолятов внутри локальных очагов сальмонеллеза) с данными кластеризации изолятов на основе результатов полногеномного секвенирования; демонстрации пространственно-временных особенностей кластеризации изолятов, относящихся к эпидемиологически не связанным случаям групповой заболеваемости.
Полученные данные впервые позволили воспроизвести детальную популяционную структуру высококлонального доминирующего серотипа сальмонелл – S. Enteritidis, являющегося превалирующим этиологическим агентом вспышечной и спорадической заболеваемости сальмонеллезами в Российской Федерации. Представленные результаты филогенетической реконструкции могут являться отправной точкой при проведении сравнительных геномных исследований в будущем, позволяя оценить направленность и динамику изменений структуры популяции S. Enteritidis.
Продемонстрирована практическая ценность геномных данных, выраженная прежде всего в высокой эпидемиологической конкордантности, наряду с высокой дифференцирующей способностью. Сравнительный анализ полногеномного секвенирования и PFGE-типирования свидетельствует о недостаточной эффективности метода PFGE для субтипирования такого высококлонального патогена, как S. Enteritids, из-за недостаточной разрешающей способности и, как следствие, его низкой информативности для определения уникальных генетических характеристик вспышечного штамма. Аналогичная проблема была описана в других работах при исследованиях изолятов S. Enteritidis [9].
Случаи выявления изолятов, принадлежащих к одному t5-кластеру на удаленных территориях, свидетельствуют о необходимости внедрения полногеномного генотипирования как при расследовании отдельных очагов групповой заболеваемости, так и при проведении мониторинга спорадических случаев и случаев изоляции сальмонелл из продуктов питания в качестве основного инструмента при поиске централизованного резервуара инфекции. Более того, данные нашего исследования свидетельствуют о случаях возникновения повторных локальных очагов заболеваний, вызванных тем же клоном S. Enteritidis на протяжении длительного временного интервала.
Результаты исследования позволили выявить некоторые особенности взаимосвязи случаев локальной вспышечной и спорадической заболеваемости сальмонеллезами на территории Российской Федерации, которые были известны ранее, но не имели фактической доказательной базы. Локальные очаги групповой заболеваемости и случаи изоляции спорадических случаев на удаленных расстояниях и в разное время, вероятно, могут являться частью более обширного эпидемического процесса, причиной которого может быть централизованное поступление контаминированной сальмонеллой продукции в разные регионы страны.
Заключение
Таким образом, выявленные в нашей работе особенности генетической кластеризации изолятов S. Enteritidis создают предпосылки к созданию единой национальной лабораторно-информационной системы по мониторингу и выявлению сходных генотипов S. Enteritidis с целью предотвращения распространения эпидемически значимых клонов патогена, а также создают основу для разработки эффективных противоэпидемических мероприятий в случае выявления эпидемически значимых клонов патогена в птицеводческих хозяйствах или предприятиях по централизованному производству продуктов питания.


